 Microglia and extracellular vesicles
Microglia and extracellular vesicles
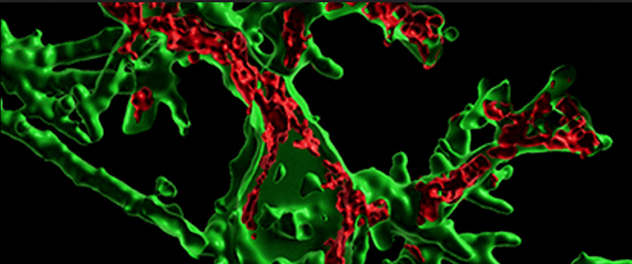
Microscopy imaging shows microglia-containing (green) extracellular vesicles (red).
Enrichment and characterization of extracellular vesicles for neuroscience research
We have established new methods for isolating extracellular vesicles from human and animal biospecimens. We also have established a protocol for profiling extracellular vesicle cargo proteins using high-sensitivity liquid chromatography and mass spectrometry.
This work has significantly advanced our technology and protein database for understanding the functional roles of extracellular vesicles, such as the role of astrocytic extracellular vesicles in neuronal differentiation and firing and diagnostic potential of brain-derived extracellular vesicles to detect Alzheimer's disease.
We also identified ATP1A3 as a neuron-specific extracellular vesicle marker. ATP1A3 can be detected in plasma and has the potential to diagnose Alzheimer's disease and related conditions.
References
- You Y, Zhang Z, Sultana N, Ericsson M, Martens YA, Sun M, Kanekiyo T, Ikezu S, Shaffer SA, Ikezu T. ATP1A3 as a target for isolating neuron-specific extracellular vesicles from human brain and biofluids. Science Advances. 2023; doi:10.1126/sciadv.adi3647.
- You Y, Muraoka S, Jedrychowski MP, Hu J, McQuade AK, Young-Pearse T, Aslebagh R, Shaffer SA, Gygi SP, Blurton-Jones M, Poon WW, Ikezu T. Human neural cell type-specific extracellular vesicle proteome defines disease-related molecules associated with activated astrocytes in Alzheimer's disease brain. Journal of Extracellular Vesicles. 2022; doi:10.1002/jev2.12183.
- Muraoka S, DeLeo AM, Sethi MK, Yukawa-Takamatsu K, Yang Z, Ko J, Hogan JD, Ruan Z, You Y, Wang Y, Medalla M, Ikezu S, Chen M, Xia W, Gorantla S, Gendelman HE, Issadore D, Zaia J, Ikezu T. Proteomic and biological profiling of extracellular vesicles from Alzheimer's disease human brain tissues. Alzheimer's & Dementia. 2020; doi:10.1002/alz.12089.
- You Y, Borgmann K, Edara VV, Stacy S, Ghorpade A, Ikezu T. Activated human astrocyte-derived extracellular vesicles modulate neuronal uptake, differentiation and firing. Journal of Extracellular Vesicles. 2019; doi:10.1080/20013078.2019.1706801.
Learn more
Learn more about our work.