Cardiac Channelopathies
Researchers in the Sudden Death Genomics Laboratory, led by Dr. Ackerman, have made numerous discoveries about cardiac channelopathies related to long QT syndrome and catecholaminergic polymorphic ventricular tachycardia.
Areas of sudden death study include:
- Genotype-phenotype relationships in long QT syndrome
- New gene discovery in the pathogenesis of long QT syndrome
- Catecholamine provocation testing in the evaluation of congenital long QT syndrome
- Sleep and neural circulatory control in long QT syndrome
- Genotype-phenotype relationships in catecholaminergic polymorphic ventricular tachycardia
Genotype-phenotype relationships in long QT syndrome
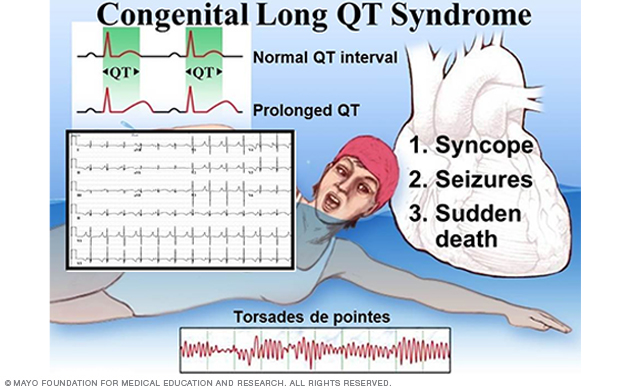
Figure 1. Clinical characteristics of long QT syndrome.
Congenital long QT syndrome (LQTS) comprises a distinct group of cardiac channelopathies characterized by delayed repolarization of the myocardium, QT prolongation and increased risk of syncope, seizures and sudden cardiac death in the setting of a structurally normal heart and otherwise healthy person (Figure 1).
This repolarization abnormality usually is without consequence. However, infrequently, when caught off guard by such triggers as exertion, swimming, emotion or auditory stimuli, the heart can spiral out of control electrically into a potentially life-threatening and sometimes lethal dysrhythmia.
The heart's rhythm typically returns to normal spontaneously after an episode of syncope. But in 5% of untreated and unsuspected cases of LQTS, the person dies of a fatal arrhythmia as the sentinel event.
Long QT syndrome is a genetically heterogeneous disorder most often inherited in an autosomal dominate mode.
To date, hundreds of mutations have been identified, with approximately 75% of clinically robust LQTS cases due to mutations in three genes — KCNQ1 (LQT1), KCNH2 (LQT2), SCN5A (LQT3) — encoding for critical cardiac ion-channel subunits that are responsible for the orchestration of the cardiac action potential (Figure 2).
Since diagnosing his first patient with long QT syndrome and carving out a postdoctoral fellowship in molecular genetics to identify that patient's LQTS-causing mutation (Pediatric Research 1998), Dr. Ackerman has investigated the genetic basis of more than 600 unrelated patients and more than 1,500 family members referred for LQTS research-based genetic testing.
The comprehensive and systematic investigation of 541 unrelated patients through all of the known LQTS-susceptibility genes (Heart Rhythm 2005 and JACC 2006) as well as the meticulous search for genetic variation among nearly 1,000 healthy volunteers from across four different ethnicities (Mayo Clinic Proceedings 2004 and Heart Rhythm 2005) has facilitated the translation of this research-based genetic test to a clinical diagnostic test called Familion, which has been available through PGxHealth since May 2004.
Moreover, research efforts in the Sudden Death Genomics Laboratory have yielded a large cohort of patients with strong clinical evidence of long QT syndrome but a negative genetic test, providing an enriched cohort for novel gene discovery.
Since the maturation of LQTS genetic testing to a commercially available molecular diagnostic test, Dr. Ackerman's research team has focused on the interpretation of genetic test results and so-called genetic purgatory with respect to the variant of uncertain and unknown significance issue.
In 2004, Dr. Ackerman's research team performed the first reverse experiment.
In this reverse experiment, instead of simply checking 50 to 200 controls to see if they did or did not possess the variant of interest (which was the standard at that time), the research team comprehensively sequenced more than 1,300 controls through the entirety of the known LQTS genes' open reading frames.
One finding of this reverse experiment was that 2% of Caucasians and 4% to 6% of non-Caucasians have rare missense variants in the SCN5A-encoded Nav1.5 sodium channel.
During the past decade, the Sudden Death Genomics Lab has been exposing this background noise issue in LQTS genetic testing and in testing for other cardiac channelopathies and cardiomyopathies.
Because the three most common LQTS-susceptibility genes had been discovered already, Dr. Ackerman initially conducted genotype-phenotype association studies involving these known genes.
This work on the known genes established swimming as a relatively LQT1-specific trigger (Mayo Clinic Proceedings 1999 and Circulation 2004) and the postpartum period as a relatively LQT2-specific temporal period for women with long QT syndrome (Heart Rhythm 2004). (Figure 2.)
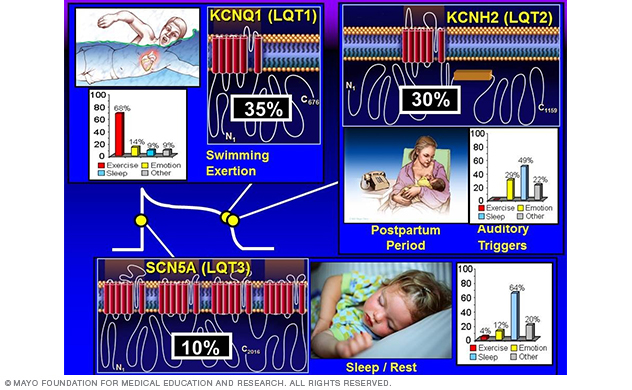
Figure 2. Genotype-phenotype correlations in long QT syndrome.
New gene discovery in the pathogenesis of long QT syndrome
Dr. Ackerman's sudden death research work has focused on elucidating novel pathogenic substrates for long QT syndrome and other cardiac channelopathies.
Since the sentinel discoveries of the first three LQTS-susceptibility genes (KCNQ1, KCNH2 and SCN5A), there have now been a total of 17 LQTS-susceptibility genes identified (Figure 3), with seven genes (Figure 3, Figure 4) discovered by Dr. Ackerman's research team (Figure 3, Figure 4).
The majority of the genes encode for ion channel interacting proteins (ChIPs), which, when mutated, functionally perturb the potassium, sodium or calcium channel pore-forming alpha subunits that are critical to orchestrating the action potential of the heart.
While many of these discoveries were made in the era of novel candidate gene analysis, the Sudden Death Genomics Lab's LQTS disease-gene discovery program is now focused on performing pedigree-based whole-exome sequencing (WES) and whole-genome sequencing (WGS).
In fact, the lab's most recent discovery of TRDN-encoded triadin (Trdn) as a novel genetic basis for recessively inherited long QT syndrome, termed triadin knockout (TKO) syndrome, was made in 2015 (Circulation 2015) using a WES-based familial genomic triangulation approach to new gene discovery (Figure 5).
Nearly 15% of the lab's overall and 50% of the lab's childhood-aged (≤ 10 years) genetically elusive LQTS cohort hosted homozygous/compound heterozygous TRDN frame-shift mutations.
A homozygous p.D18fs*13 mutation was identified in an African-American girl (Figure 6). The same exact homozygous p.K147fs0* mutation was identified in three unrelated patients of either Indian or Arabic descent. And one Caucasian male was found to be compound heterozygous for two frame-shift mutations — p.N9fs*5 and p.K147fs*0.
Since frame-shift mutations often result in nonfunctional proteins or immediate nonsense-mediated RNA decay, these patients are expectantly Trdn null.
Remarkably, all five Trdn null children displayed the common electrocardiographic phenotype of extensive T-wave inversions in precordial leads V1-V4, with either persistent or transient QT-prolongation, severe disease expression of exercise-induced cardiac arrest in early childhood, a recessive inheritance pattern, and required aggressive therapy.
Patients often presented with sudden cardiac arrest before the age of 5, and current therapeutic strategies (such as beta blockers and left cardiac sympathetic denervation surgery) have been ineffective.
The research team in the Sudden Death Genomics Lab is now in the process of further characterizing TKO syndrome using patient-specific induced pluripotent cell derived cardiomyocytes (iPSC-CMs).

Figure 3. Summary of LQTS-susceptibility genes. The yellow boxes show genes discovered by the Sudden Death Genomics Laboratory.
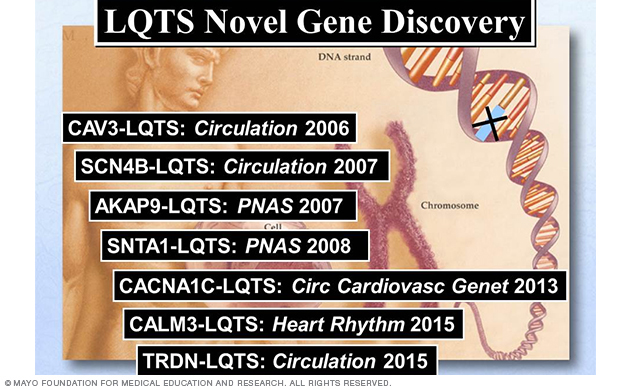
Figure 4. Summary of LQTS-susceptibility genes discovered by Dr. Ackerman's Sudden Death Genomics Laboratory at Mayo Clinic.
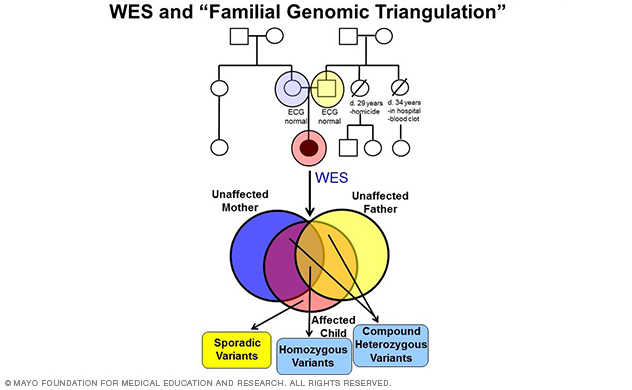
Figure 5. A depiction of whole-exome sequencing and familial genomic triangulation-based new gene discovery.
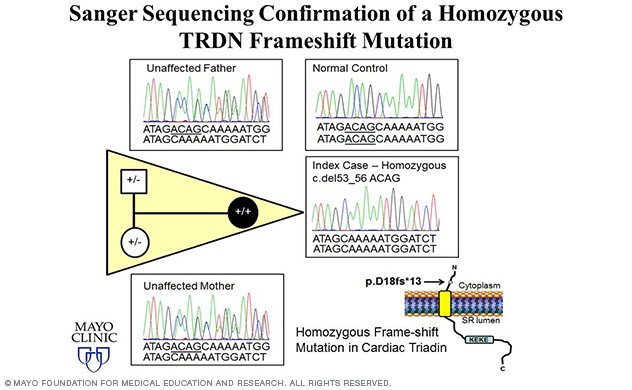
Figure 6. Elucidation of triadin knockout syndrome.
Catecholamine provocation testing in the evaluation of congenital long QT syndrome
Determining the influence of various stimuli (such as auditory arousal from sleep and activation of dive reflex) on muscle sympathetic nerve activity and cardiac repolarization are the principal pursuits with respect to these human LQTS studies.
Through this patient-oriented research, the Sudden Death Genomics Lab has developed the Mayo Epinephrine QT Stress Test (Mayo Clinic Proceedings 2002) and demonstrated that paradoxical lengthening of the absolute QT interval during low-dose epinephrine infusion has 75% positive predictive value and 96% negative predictive value with respect to type 1 LQTS (Circulation 2006; Figure 7).
This clinical diagnostic test is now used in heart rhythm centers throughout the world in an effort to unmask patients with concealed LQT1.
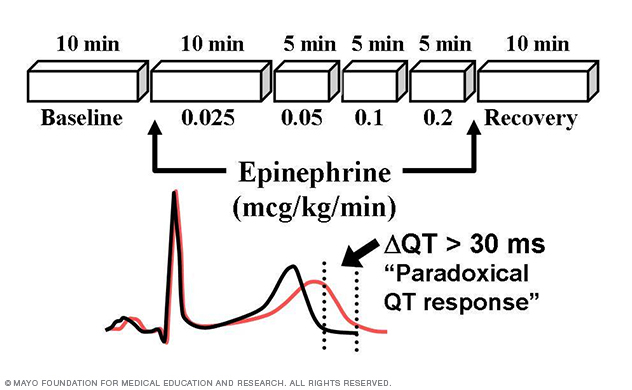
Figure 7. Long QT syndrome stress test.
Sleep and neural circulatory control in long QT syndrome
More than 200 patients with genotype-positive LQTS have been research participants in the lab's studies investigating neural circulatory control in LQTS (HL/NS70302).
Overnight sleep studies identified dysregulated sleep architecture and abnormal cardiac repolarization in women with type 2 LQTS (Circulation 2002).
Microneurography studies revealed profoundly attenuated muscle sympathetic nerve activity in all patients with long QT syndrome regardless of the mutated gene (Circulation 2003).
Genotype-phenotype relationships in catecholaminergic polymorphic ventricular tachycardia
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is another heritable arrhythmia syndrome that classically manifests with exertion-induced syncope or sudden death and closely mimics that of long QT syndrome.
Catecholaminergic polymorphic ventricular tachycardia is associated with a completely normal resting electrocardiogram (ECG) and is electrocardiographically suspected after either exercise or catecholamine stress testing that demonstrates significant ventricular ectopy.
Similar to LQTS, catecholaminergic polymorphic ventricular tachycardia is generally associated with a structurally normal heart.
Perturbations pursuant to mutations in the RyR2¬-encoded cardiac ryanodine receptor represent the most common genetic subtype of CPVT (Figure 8). About 65% of CPVT is due to RyR2 mutations and is regarded as type 1 CPVT (CPVT1).
The lethality of CPVT is illustrated by the presence of a positive family history of juvenile (< 40 years) sudden cardiac death for more than one-third of people with catecholaminergic polymorphic ventricular tachycardia and in as many as 60% of families hosting RyR2 mutations.
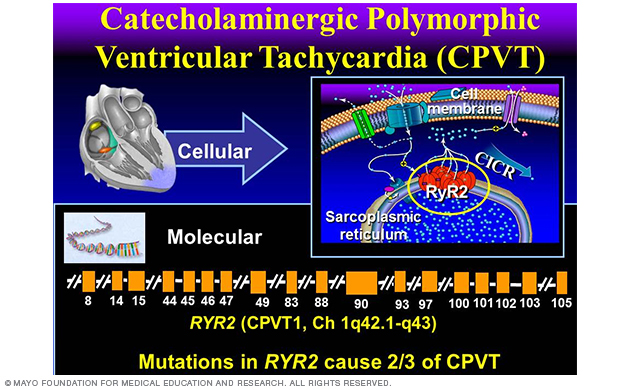
Figure 8. Catecholaminergic polymorphic ventricular tachycardia.