Research
Ongoing research in Dr. Petrucelli's Neurodegenerative Diseases Lab centers on cellular mechanisms induced by the abnormal aggregation of specific proteins and their contribution to neuronal dysfunction and death.
Modeling c9FTD/ALS in cells and mice
A hexanucleotide repeat expansion in a noncoding region of the gene chromosome 9 open reading frame 72 (C9orf72) is the most common genetic cause of frontotemporal dementia and amyotrophic lateral sclerosis (ALS). As these two conditions can occur in the same person, they are considered to be a disease spectrum referred to as c9FTD/ALS. Since the identification of this disease spectrum in 2011, our lab has been at the forefront of research on this condition.
The lab contributed to the initial characterization of the repeat-associated non-ATG (RAN) translation of this repeat, which results in the formation of five different dipeptide repeat (DPR) proteins. We generated the first mouse model to recapitulate the various pathological features associated with c9FTD/ALS and developed subsequent mouse models that express longer repeat expansions and individual DPR proteins. We continue to explore the role of DPR proteins in disease pathogenesis and seek to apply our findings to the development of new therapeutics.
Progranulin, neuroinflammation and toxicity
Mutations in the granulin (GRN), which result in a loss of the protein progranulin (PGRN), are associated with TAR DNA-binding protein 43 (TDP-43)-positive frontotemporal dementia. PGRN plays a role in several cellular processes, including cell growth, neuronal repair and inflammation. In its full-length form, the protein has trophic and anti-inflammatory functions; yet, PGRN also can be cleaved into smaller fragments that have proinflammatory properties.
Our lab and others have found that PGRN can be cleaved by the lysosomal enzyme cathepsin D, and Grn knockout mice develop lysosomal abnormalities. These mice also show aberrant microglial activation, suggesting an interesting link between lysosomal dysfunction and immune responses. Neuroinflammatory pathways have long been suspected to influence neuronal degeneration, and our lab uses both mouse models and cell biology to investigate the interplay between these different disease-associated processes.
 PGRN levels
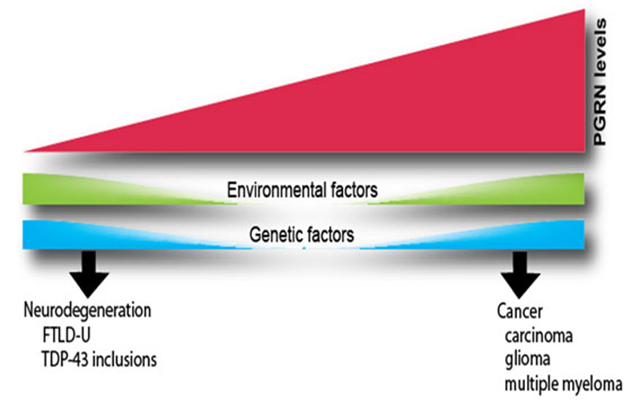
PGRN levels
Changes in the expression of the PGRN affect many biological functions. Elevated PGRN levels stimulate proliferation, survival and motility in a variety of cells. As a result, PGRN acts as a tumorigenic agent in several types of cancers. Conversely, genetic changes that result in lower PGRN levels are a major cause of a subtype of frontotemporal dementia, the ubiquitinated type with TDP-43-positive inclusions (FTLD-U). Variations in the clinical presentation of FTLD-U, particularly the observation that disease onset can occur over a broad range of ages, suggest that other factors also influence PGRN levels in humans. Such factors could be environmental or genetic.
The effects of TDP-43 loss on RNA processing
As TDP-43 normally acts to repress the inclusion of cryptic exons in RNA transcripts, the mislocalization and aggregation of TDP-43 results in an increase in cryptic exon-containing transcripts in the tissues of people with ALS and frontotemporal dementia. With the help of our collaborators, we are identifying which TDP-43 targets are aberrantly spliced in the disease state and developing tools to detect the resulting cryptic peptides in vivo.
Our research team suspects that measuring the levels of these cryptic peptides could act as a proxy for measuring TDP-43 pathology in patient samples. Furthermore, should the levels of any cryptic peptide correlate with clinical characteristics such as age of onset or disease duration, that peptide could be used as a prognostic biomarker in ALS and frontotemporal dementia. We also are assessing whether any of these misspliced products contribute to disease pathogenesis using cell and animal models. Our investigators believe that this line of inquiry will open new avenues of research on TDP-43 biology and frontotemporal dementia and ALS pathogenesis.
Measuring tau seeding and its role in disease progression
The accumulation of hyperphosphorylated tau causes the formation of neurofibrillary tangles, a pathological hallmark of Alzheimer's disease, frontal temporal dementia, progressive supranuclear palsy, Pick's disease, corticobasal degeneration and other tauopathies. Understanding the variables that influence the rate and spread of tau aggregation could profoundly affect the treatment of these complex disorders.
Recently, our team learned that a smaller part of the abnormal tau protein present in people with Alzheimer's disease has the intrinsic ability to polymerize under physiological conditions. This region is referred to as the Alzheimer's disease tau core. Furthermore, the Alzheimer's disease tau core can template wild-type tau, inducing its incorporation into tau aggregates. Using an ultrasensitive real-time quaking-induced conversion (RT-QuIC) assay, our lab can measure the inherent seeding ability of abnormal tau species in patient samples. We hope that the use of this assay will allow us to gain diagnostic and prognostic insight in Alzheimer's disease, and we aim to apply these methods to other tauopathies as well.
Biomarker assay development
Across all of our lab's research, our team is diligently looking for factors that can be exploited as biomarkers. We then use our expertise in immunoassay development to track the levels of various proteins in patient tissues, cerebrospinal fluid and blood. Using this approach, we have analyzed the levels of disease-specific proteins and peptides in multiple conditions, including ALS, frontotemporal dementia and spinocerebellar ataxia 3.
Our lab also has developed assays to measure neurofilament levels in patient samples. Neurofilaments accumulate in patient biofluids upon axonal degeneration and can thereby act as a measure of neuronal death. We observe changes in neurofilament levels in people with several diseases that we study, as well as in patients with stroke and, most recently, those hospitalized with COVID-19.