Mutations in Epigenetic Regulator Genes as Drivers of Cancer
 Epigenetic modification at the histone H3 lysine 36 position.
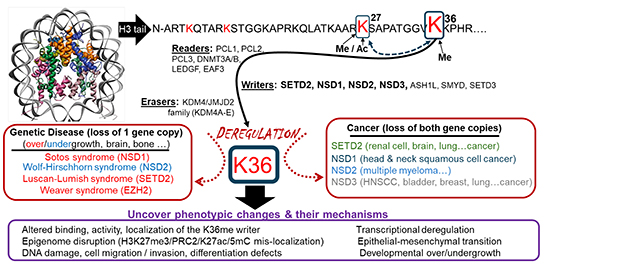
Epigenetic modification at the histone H3 lysine 36 position.
This diagram illustrates epigenetic writers, readers and erasers of histone H3 lysine 36 methylation, their links to human disease and cancer, and the cellular phenotypes they regulate. These regulators are studied in the contexts of cancer and developmental syndromes to better understand how these conditions arise and to find new ways to treat people affected by them.
Genes regulating epigenetic marks mutate across a broad spectrum of human cancers. For example, the ten-eleven translocation (TET) family of proteins mutates in nearly 30% of myeloid malignancies overall. The TET family also mutates in up to 60% in certain subtypes of these malignancies, such as chronic myelomonocytic leukemia. Histone methyltransferase SETD2 mutates in about 25% of primary clear cell renal cell cancers and over 60% of clear cell renal cell cancer metastases.
How these mutations promote cancer and metastasis is likely distinct from the mechanism of traditional tumor suppressor genes, such as p53. This difference demands a better mechanistic understanding of how these mutations regulate cell growth, differentiation and response to therapy. Elucidating their mechanism of action also is expected to yield novel vulnerabilities created by these mutations that can be developed into new targeted therapies.
To study this process, the Epigenetic Etiology of Human Disease Laboratory is developing cell culture, organoid and animal model systems to probe the impact of several commonly occurring epigenetic regulator gene mutations. The lab will probe at the epigenome and transcriptome levels, at single genes, and over the entire genome. At present, these genes include TET2, DNMT3A and SETD2, as well as the NSD1, NSD2 and NSD3 family.
Renal cell cancer — one of the focus areas of the Epigenetic Etiology of Human Disease Laboratory — is distinctive because it is heavily dominated by epigenetic regulator gene mutations, including SETD2, PBRM1 and BAP1, while presenting with relatively low mutation frequency of traditional tumor suppressor genes such as p53. SETD2 is a histone methylase that trimethylates the histone H3 lysine 36 (H3K36) position, a mark enriched in the bodies of actively transcribed genes. Interestingly, H3K36me3 recruits DNMT3A and DNMT3B to promote DNA methylation within gene bodies. In some regions of the genome, H3K36me3 antagonizes the function of polycomb repressive complex 2 (PRC2), which writes the H3K27 trimethylation mark to silence gene expression.
Our laboratory showed that mutations in SETD2 globally deregulate DNA methylation patterns and create a DNA hypermethylator phenotype that may promote more-aggressive forms of clear cell renal cell cancer. In other studies using CRISPR knockout screens, we discovered a synthetic lethal relationship between SETD2 and NSD1, thus revealing that pharmacological targeting of NSD1 could serve as an individualized treatment for SETD2 loss-of-function cancers. By understanding the mechanisms underpinning epigenetic regulator mutations such as SETD2, we can better understand how they drive tumor initiation and progression and impact patient survival and response to treatment.
These studies in the Epigenetic Etiology of Human Disease Laboratory also are expected to yield novel and personalized treatments for people with cancer presenting with mutations in epigenetic regulator genes. The research team hypothesizes that tumors with epigenetic mutations will be more susceptible to drugs that target the epigenome, creating new avenues for individualized therapy.