Research Projects
Research projects in the Cholangiopathies Laboratory focus on complex liver diseases that currently have limited treatment options. Dr. LaRusso's ultimate goal is to expand treatment options for patients by advancing knowledge about primary sclerosing cholangitis and polycystic liver diseases.
Primary sclerosing cholangitis
Primary sclerosing cholangitis
Primary sclerosing cholangitis
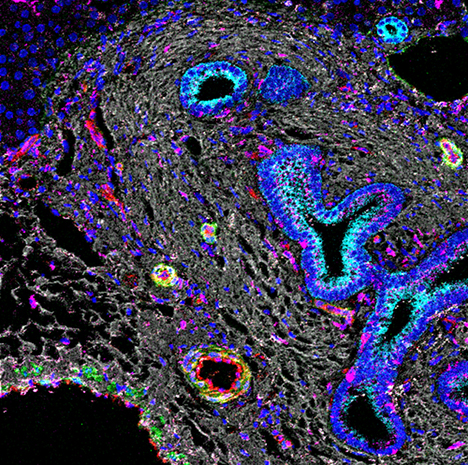
Primary sclerosing cholangitis
In this image, imaging mass cytometry shows liver tissue from a patient with primary sclerosing cholangitis. The cyan color shows positivity for cytokeratin. The magenta color shows an infiltration of macrophages.
Primary sclerosing cholangitis is a rare, incurable, complex autoimmune disease of the bile ducts, with a median survival time of 13 to 20 years. Primary sclerosing cholangitis affects about 30,000 Americans, accounts for about 6% of liver transplants and incurs about $125 million in health care costs every year.
Our research team has proposed that primary sclerosing cholangitis likely develops and progresses through the interaction of predisposing genetic factors and environmental exposures. This interaction influences biological processes that occur at the level of the gut and the biliary tree, including the intestinal microbiome, and through dynamic changes in cholangiocyte phenotype, including its epigenome.
Research from our lab showed that in patients with primary sclerosing cholangitis, cholangiocytes undergo the process of cellular senescence, a phenotype linked to both cell cycle arrest and vigorous secretion of various bioactive molecules — a process called the senescence-associated secretory phenotype. Cells with this phenotype can modify their microenvironment by inducing proinflammatory and fibrotic cellular reactions and accelerating neoplastic transformation.
Our work is providing mechanistic information about the pathways of senescence and senescence-associated secretory phenotype, inflammation and fibrosis in primary sclerosing cholangitis, and it has substantial therapeutic implications for the development and testing of drugs that selectively kill senescent cells (senolytics).
Polycystic liver diseases
Polycystic liver disease
Polycystic liver disease
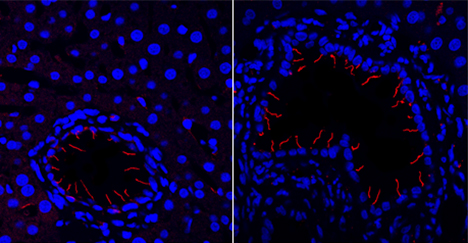
Polycystic liver disease
This image shows a rodent model of polycystic liver disease. The left panel shows no drug treatment, and the right panel shows treatment with hydroxychloroquine, which increases ciliary length.
Polycystic liver disease (PLD) is a genetic ciliopathy for which there are no regulatory approved drugs. The aims of the PLD research program within our lab are to clarify the pathogenesis of polycystic liver disease and develop effective therapies.
We have previously reported that PLD cholangiocytes (PLDCs) have increased cyclic adenosine monophosphate (cAMP), accelerated autophagy, and structurally and functionally malformed cilia. However, it remains unclear what mechanisms are involved, how they contribute to hepatic cystogenesis or if they represent new therapeutic targets.
Hepatic cysts arise from cholangiocytes exhibiting a hyperproliferative phenotype. Considering that hyperproliferation of many cell types is associated with alterations in autophagy, our current research efforts focus on this degradative process in polycystic liver disease cholangiocytes, how it contributes to hepatic cystogenesis and whether it might represent a potential therapeutic target. Our objectives are to explore the mechanistic relations between autophagy of ciliogenic proteins (that is, proteins involved in cilia maintenance), pro-cystogenic signaling and hepatic cystogenesis.
Research studies include testing the hypothesis that overexpression of TGR5, a bile acid membrane receptor, in cystic cholangiocytes induces selective autophagy of discrete molecules, including key cystogenic proteins and microRNAs (miRNAs) via the calcium and cyclic adenosine monophosphate (cAMP) signaling pathways.
To test these hypotheses, we employ functional pathway cluster analysis and next-generation sequencing, transmission and scanning electron microscopy, immunofluorescence confocal microscopy, novel methods to study ciliary dynamics, 3D culture systems of cystogenesis, microfluidic models to studying mechano-chemo ciliary sensing and signal transduction, human tissue, and disease rodent models.
Our results will be informative for mechanisms of polycystic liver disease progression and potentially transformative for new therapeutic approaches.